|
CoMD
A Mini-app for Co-Design of Classical Molecular Dynamics.
|


|
|
CoMD
A Mini-app for Co-Design of Classical Molecular Dynamics.
|
|
Leapfrog time integrator. More...
#include "CoMDTypes.h"
Go to the source code of this file.
Functions | |
| double | timestep (SimFlat *s, int n, real_t dt) |
| Advance the simulation time to t+dt using a leap frog method (equivalent to velocity verlet). More... | |
| void | computeForce (SimFlat *s) |
| void | kineticEnergy (SimFlat *s) |
| Calculates total kinetic and potential energy across all tasks. More... | |
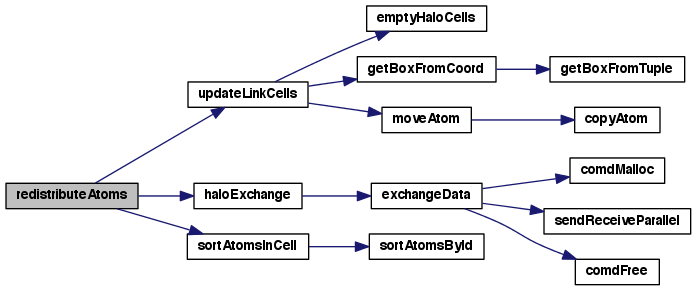
| void | redistributeAtoms (struct SimFlatSt *sim) |
| Update local and remote link cells after atoms have moved. More... | |
Leapfrog time integrator.
Definition in file timestep.h.
| void computeForce | ( | SimFlat * | s | ) |
| void kineticEnergy | ( | SimFlat * | s | ) |
Calculates total kinetic and potential energy across all tasks.
The local potential energy is a by-product of the force routine.
Definition at line 97 of file timestep.c.
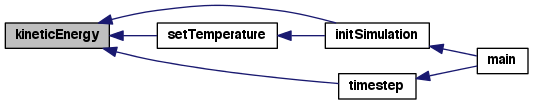
| void redistributeAtoms | ( | SimFlat * | sim | ) |
Update local and remote link cells after atoms have moved.
This function provides one-stop shopping for the sequence of events that must occur for a proper exchange of halo atoms after the atom positions have been updated by the integrator.
Definition at line 136 of file timestep.c.
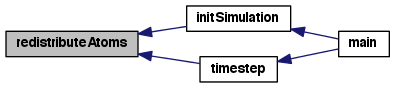
Advance the simulation time to t+dt using a leap frog method (equivalent to velocity verlet).
Forces must be computed before calling the integrator the first time.
This leaves positions, velocities, and forces at t+dt, with the forces ready to perform the half step velocity update at the top of the next call.
After nSteps the kinetic energy is computed for diagnostic output.
Definition at line 31 of file timestep.c.
 1.8.3.1
1.8.3.1